As outlined in Methods, we implemented two alternative approaches for generating synthetic frequency distributions that recapitulate either positive selection or neutral evolution. The algorithm for simulating tumours (variant allele frequency distributions) subject to positive selection (adapted from Williams et al. 2018) is outlined in Algorithm 1. The algorithm for generating neutral frequency distributions is outlined in Algorithm 2 (inspired by Caravagna et al. 2020). The complete algorithm for the paired simulation of positively selected and neutrally evolving tumours is outlined in Algorithm 3. Software to generate synthetic tumours/VAF distributions can be found on github @tomouellette/CanEvolve.jl
\begin{algorithm}
\caption{Tumours subject to positive selection (adapted from Williams et al. 2018)}
\begin{algorithmic}
\STATE \tiny Simulate tumour with $Q$ positively selected subclones with frequencies $>$ $L$ and $<$ U
\WHILE {(U $<$ subclone frequency $<$ L) and (number of subclones \NOT $Q$)}
\STATE 1. Initialize cell with $n_{clonal}$ clonal mutations
\WHILE{current population size $<$ $N$}
\STATE 2. Randomly sample a cell $j$
\STATE 3. Draw a random number $r$ from Uniform($a$, $b$) where $a$ = 0 and $b$ = $b_{max}$ + $d_{max}$ (maximum birth and death rates of all cells in population)
\STATE 4. With $r$, cell $j$ will divide with probability proportional to its birth rate $b_j$ and die with a probability proportional to its death rate $d_j$
\IF{$b_j$ $>$ $r$}
\STATE 5a. Cell divides and both daughter cells acquire $k$ mutations where $k$ is Poisson distributed with mean equal to the per genome division mutation rate $\mu$
\STATE 5i. Each mutation has a probability $P_d$ of being a positively selected driver and initiating a new subclone
\STATE 5ii. If the mutation is a driver, it is assigned a selection coefficient $s$ randomly sampled from an exponential distribution with a scale parameter 1/$\lambda$
\STATE 5iii. The time (in current population size $n$ divided by final population size $N$) is recorded for every mutation
\ELIF{$b_j$ + $d_j$ $>$ $r$ $>=$ $b_j$}
\STATE 5b. Cell dies
\ELSE
\STATE 5c. Nothing happens
\ENDIF
\ENDWHILE
\ENDWHILE
\STATE 6. Virtual biopsy synthetic tumour and add sequencing noise
\STATE 7. Remove mutations below hard alternate read cutoff (e.g 2 / mean sequencing depth)
\end{algorithmic}
\end{algorithm}
\begin{algorithm}
\caption{Neutrally evolving tumours (inspired by Caravagna et al. 2020)}
\begin{algorithmic}
\STATE \tiny Simulate a neutral variant allele frequency distribution observed in bulk sequenced tumour populations
\STATE 1. Randomly sample or set shape $\alpha$ and scale $\beta$ parameters for a Pareto distribution
\STATE 2. Generate neutral 'tail' mutations by sampling $n_{non-clonal}$ mutations from Pareto($\alpha$, $\beta$)
\STATE 3. Add $n_{clonal}$ heterozygote mutations at a frequency of 0.5
\STATE 4. With some probability $P_{trim}$, remove variants below a randomly sampled frequency $f$ (e.g. 0.1 - 0.3) to mimic the loss of neutral tails observed in empirical samples (in general, $P_{trim}$ < 0.1)
\STATE 5. Add sequencing noise
\STATE 6. Remove mutations below hard alternate read cutoff (e.g 2 / mean sequencing depth)
\end{algorithmic}
\end{algorithm}
Supplementary Figure 1. An example visualization of VAF distribution generation for neutral synthetic tumours
\begin{algorithm}
\caption{Paired simulation of VAF distributions from neutrally evolving and positively selected tumours}
\begin{algorithmic}
\STATE \tiny 1. Specify number of subclones $Q$ and minimum and maximum subclone frequencies $L$ and $U$
\STATE 2. Randomly sample simulation parameters: $\mu$ (per genome per division mutation rate), $P_d$ (probability of driver/subclone event), $n_{clonal}$, $n_{drivers}$, $\lambda^{-1}$ (scale parameter), mean sequencing depth, $\rho$ (sequencing overdispersion parameter)
\STATE 3. Run Algorithm 1 until synthetic data is generated with $Q$ subclones at frequencies $<$$U$ and $>$$L$
\STATE 4. Count the approximate number of clonal $n_{clonal}$ and non-clonal $n_{non-clonal}$ mutations in the positive selection scenario
\STATE 5. Run Algorithm 2 using $n_{clonal}$ * $\psi_{a}$ and $n_{non-clonal}$ * $\psi_{b}$ mutations. $\psi_{a}$ and $\psi_{b}$ are uniformly sampled numbers that scale the number of clonal and non-clonal mutations to capture additional heterogeneity in training sets.
\end{algorithmic}
\end{algorithm}
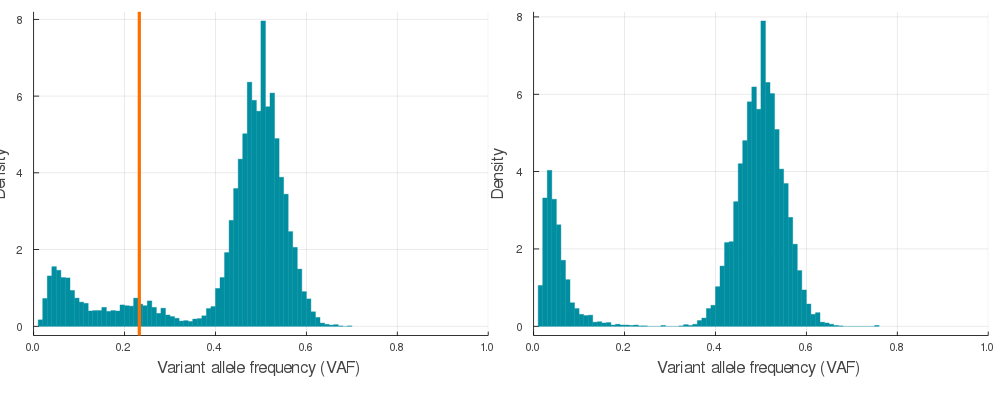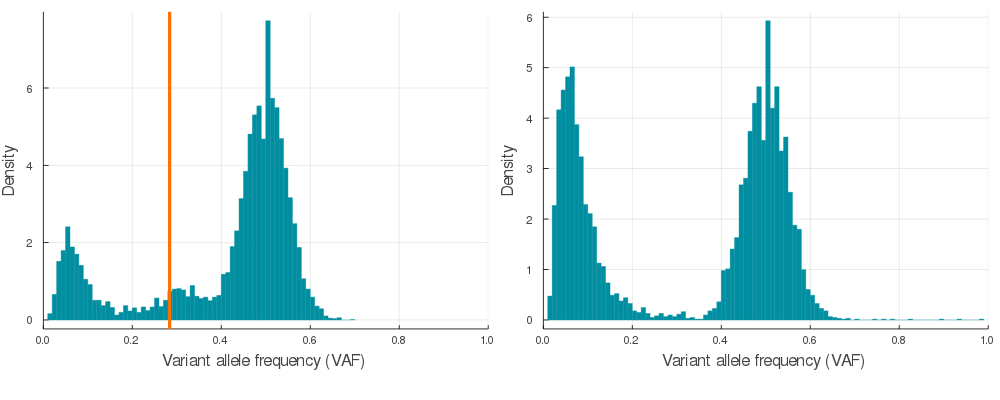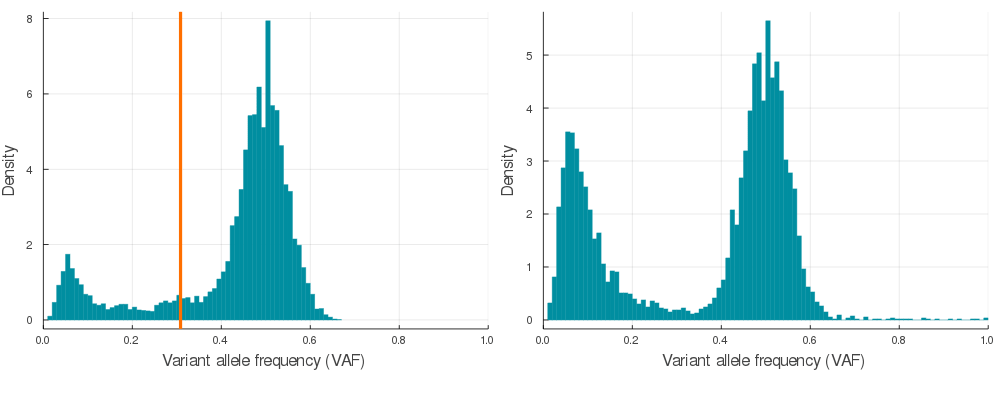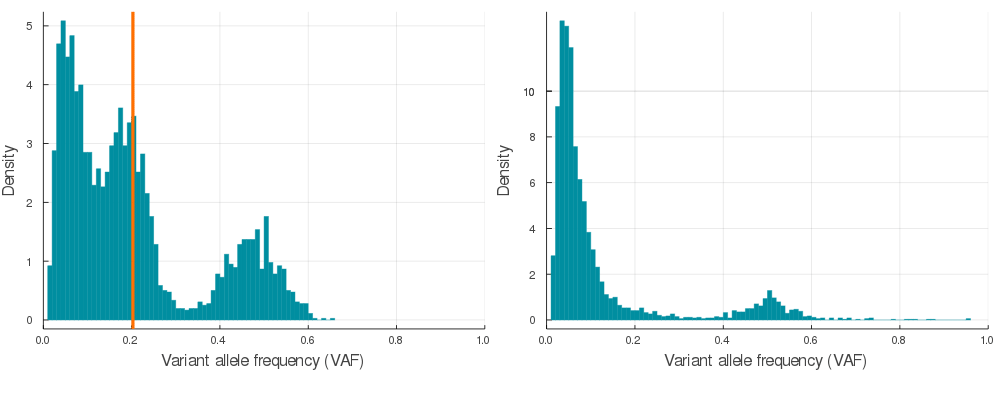
Supplementary Figure 2. A sample of 20 paired simulations with positive selection (left) and neutral evolution (right). Orange lines indicate subclone frequencies (cellular proportion / 2)